Researchers from the Centre for Endocrinology win 2019 EJE Readers’ Choice Award for joint study into rare disease
Q&A with Dr Helen Storr, Reader and Honorary Consultant in Paediatric Endocrinology
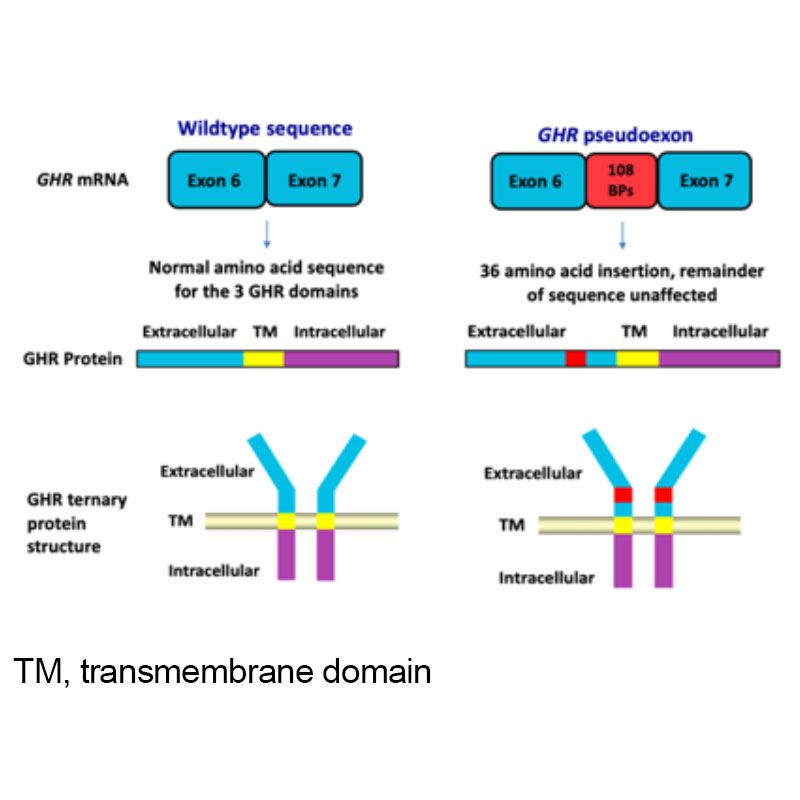
Figure 1. Growth Hormone Receptor (GHR) pseudoexon (6Ψ) mutation
What is new about the study?
A rare homozygous intronic GH receptor (GHR) pseudoexon (6Ψ) mutation was first described by our group in 2001 (Figure 1). This novel genetic abnormality causes growth failure due to growth hormone insensitivity (GHI) / IGF-1 deficiency. We have subsequently identified / genetically diagnosed a number of other UK children with this disorder.
This novel patient cohort has proven highly informative as it provides insights into the spectrum of GHI disorders. A genetic diagnosis of GHI is critical in patient management as it is now clear that recombinant human IGF-1 therapy (rhIGF-1) is more likely to achieve height gain than standard GH therapy.
Our work details the wide phenotypic variability of twenty patients with GHR 6Ψ mutations and we believe it significantly enhances understanding of the expanding spectrum of GHI defect subtypes. Although rhIGF-1 replacement therapy has been evaluated in children with severe IGF-1 deficiency, its efficacy in less severe GHI patients is unclear. We report for the first time, the response to rhIGF-1 therapy in this cohort.
Was there anything surprising about the results?
Homozygous intronic 6Ψ GHR mutations causes both severe and milder (‘non-classical’) GHI phenotypes, even in individuals within the same kindred. The presence of abnormal facial features did not correlate with either the degree of short stature or with biochemical abnormalities. This is in contrast to patients with other molecular causes of GHI, where facial anomalies indicate more severe disease forms.
Previous studies in GHR mutation patients have suggested that the height deficiency correlates with degree of IGF-1 deficiency. Conversely, we found no such correlation between the clinical (degree of short stature) and biochemical (IGF-1 deficiency) features in individual patients.
In our cohort, even those individuals with milder forms of IGF-1 deficiency, rhIGF-1 treatment improved long-term height, demonstrating responses similar to those patients with more severe GHI / primary IGF1 deficiency (Figure 2).
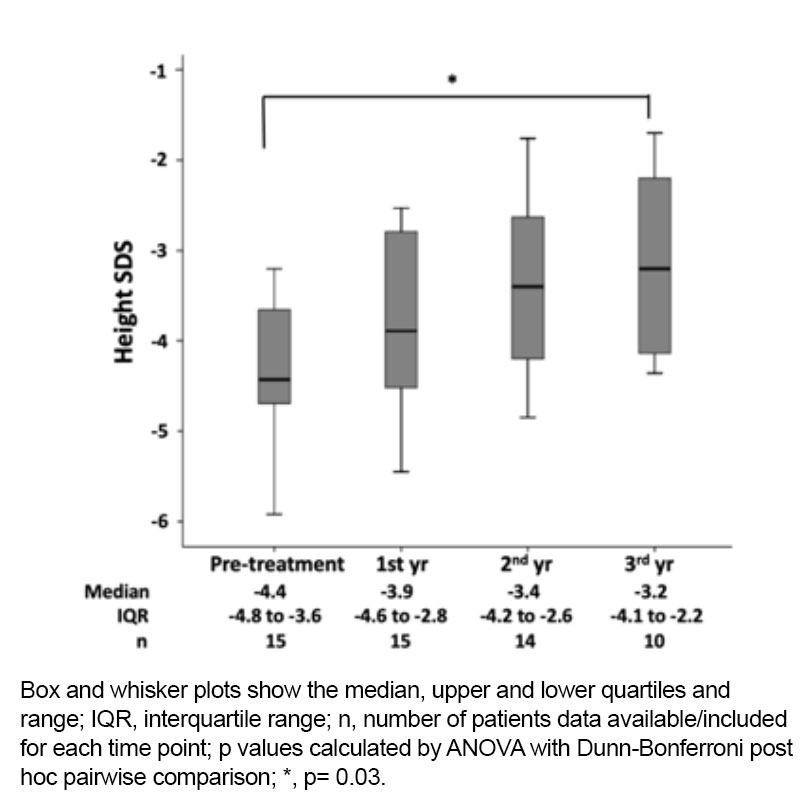
Figure 2. Height SDS at four different time points during treatment with recombinant human IGF-1 therapy.
Why is the study important and what are the wider implications?
Integrity of the GH-IGF-1 axis is fundamental for normal human growth. Defects in several genes of this axis are recognised to cause GHI / severe childhood growth impairment. Detailed clinical, biochemical, and genetic characterisation studies of patients with GHI phenotypes are important. These studies enhance our understanding of GHI and overlapping short stature syndromes and also elucidate the cellular mechanisms underpinning normal human growth.
Milder GHI phenotypes form a clinically important subgroup and prevalence may be much higher than ‘classical’ more severe GHI. Our work is particularly important in highlighting the clinical and biochemical features, particularly of phenotypes at the milder end of the GHI spectrum.
Finally, this study provides encouraging preliminary data on treatment responses and helps guide clinicians managing this challenging condition. Our key message to clinicians and scientists is that patients with mild to moderate GHI should be investigated and considered for treatment.
My team and I are honoured to receive the prestigious EJE Readers’ choice award. We are delighted that the readership of this leading endocrine journal confirms our belief that this work is of relevance and interest to the broader endocrine community. Although this is a rare disease, it has a significant clinical impact.
Further information
- Research paper: Phenotypic spectrum and responses to recombinant human IGHF1 (rhIGF1) therapy in patients with pseudoexon growth hormone receptor mutation by Sumana Chatterjee et al, European Journal of Endocrinology, doi.org/10.1530/EJE-18-0042
- Find out more about Endocrinology research at the William Harvey Research Institute